
بیماری کلیه پلی کیستیک ( به انگلیسی: Polycystic kidney disease ) یا اختصاراً PKD یک بیماری مادرزادی کلیه است که در آن دو کلیه دارای کیست های متعدد هستند.
این بیماری ژنتیکی از شایع ترین بیماری های مادرزادی کلیه است ( حدود دوازده و نیم میلیون بیمار در سراسر جهان ) و دو گونه دارد نوع اتوزومال غالب ( ADPKD ) و نوع اتوزومال مغلوب ( ARPKD ) ، که نوع اول شایع تر است. بیماری ADPKD می تواند در پی وقوع جهش در ژن های PKD - 1 ( کروموزوم 16 ) ، PKD - 2 ( کروموزوم4 ) و یا احتمالاً PKD3 رخ دهد.
در بیش از هشتاد درصد موارد کیست های متعدد مملو از مایع در هردو کلیه مشاهده می شوند ولی گاه کیست ها فقط در یک طرف وجود دارند. کیست ها می توانند در کبد و لوزالمعده و به ندرت قلب نیز مشاهده شوند.
در نوع اتوزومال غالب هرچند تشکیل کیست ها از دوران جنینی آغاز می شود ولی تا جوانی اغلب بیماران بدون علامتند ؛ علائم بتدریج ظاهر می شوند مانند افزایش فشارخون، درد پهلو، توده شکمی ( ناشی از بزرگ شدن کلیه ) ، تهوع، خستگی، عفونت های مکرر ادراری و هماچوری آشکار یا میکروسکوپیک که به درمان علامتی نیاز دارند. با این حال بیماری ماهیتی پیشرونده دارد و غالباً تا دهه چهارم یا پنجم زندگی منجر به بیماری کلیوی مرحله نهایی ( End Stage Renal Disease = ESRD ) می شود.
تا قبل از بروز ESRD درمان علامتی است مانند وازوپرسین و Tolvaptan. پس از بروز نارسایی کلیه در مراحل انتهایی درمان دیالیز و پیوند کلیه است. ده درصد بیماران دیالیزی ( مبتلایان به نارسایی کلیه ) را این بیماران تشکیل می دهند. در این بیماران به دلیل بزرگی کلیه گاه قبل از پیوند کلیه نیاز به نفرکتومی است. درمان با سلول های بنیادی در مراحل تحقیقاتی است.
بیماری نوع مغلوب سیر سریع تری دارد و اغلب در نوزادی علائم بروز می کنند.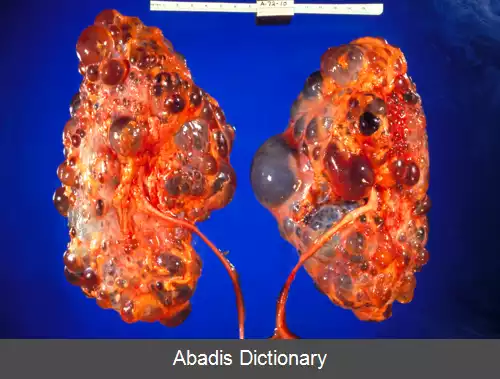
این نوشته برگرفته از سایت ویکی پدیا می باشد، اگر نادرست یا توهین آمیز است، لطفا گزارش دهید: گزارش تخلفاین بیماری ژنتیکی از شایع ترین بیماری های مادرزادی کلیه است ( حدود دوازده و نیم میلیون بیمار در سراسر جهان ) و دو گونه دارد نوع اتوزومال غالب ( ADPKD ) و نوع اتوزومال مغلوب ( ARPKD ) ، که نوع اول شایع تر است. بیماری ADPKD می تواند در پی وقوع جهش در ژن های PKD - 1 ( کروموزوم 16 ) ، PKD - 2 ( کروموزوم4 ) و یا احتمالاً PKD3 رخ دهد.
در بیش از هشتاد درصد موارد کیست های متعدد مملو از مایع در هردو کلیه مشاهده می شوند ولی گاه کیست ها فقط در یک طرف وجود دارند. کیست ها می توانند در کبد و لوزالمعده و به ندرت قلب نیز مشاهده شوند.
در نوع اتوزومال غالب هرچند تشکیل کیست ها از دوران جنینی آغاز می شود ولی تا جوانی اغلب بیماران بدون علامتند ؛ علائم بتدریج ظاهر می شوند مانند افزایش فشارخون، درد پهلو، توده شکمی ( ناشی از بزرگ شدن کلیه ) ، تهوع، خستگی، عفونت های مکرر ادراری و هماچوری آشکار یا میکروسکوپیک که به درمان علامتی نیاز دارند. با این حال بیماری ماهیتی پیشرونده دارد و غالباً تا دهه چهارم یا پنجم زندگی منجر به بیماری کلیوی مرحله نهایی ( End Stage Renal Disease = ESRD ) می شود.
تا قبل از بروز ESRD درمان علامتی است مانند وازوپرسین و Tolvaptan. پس از بروز نارسایی کلیه در مراحل انتهایی درمان دیالیز و پیوند کلیه است. ده درصد بیماران دیالیزی ( مبتلایان به نارسایی کلیه ) را این بیماران تشکیل می دهند. در این بیماران به دلیل بزرگی کلیه گاه قبل از پیوند کلیه نیاز به نفرکتومی است. درمان با سلول های بنیادی در مراحل تحقیقاتی است.
بیماری نوع مغلوب سیر سریع تری دارد و اغلب در نوزادی علائم بروز می کنند.

wiki: بیماری کلیه پلی کیستیک