
مکانیک مولکولی
فرهنگستان زبان و ادب
دانشنامه عمومی
مکانیک مولکولی از مکانیک کلاسیک برای مدل سازی سیستم های مولکولی استفاده می کند. تقریب بورن - اوپنهایمر معتبر قرض می شود و انرژی بالقوه همه سیستم ها به عنوان تابعی از مختصات هسته ها با استفاده از میادین نیرو محاسبه می شود. مکانیک مولکولی می تواند برای مطالعه سیستم های مولکولی متفاوت از نظر اندازه و پیچیدگی، سیستم های بیولوژیکی کوچک تا بزرگ یا مجموعه ها یا خوشه های مواد با هزاران تا میلیون ها اتم استفاده شود.
روشهای مکانیک مولکولی اتمی دارای خصوصیات زیر است:
• هر اتم به عنوان یک ذره شبیه سازی می شود
• به هر ذره یک شعاع اختصاص داده می شود ( به طور معمول شعاع ون در والس ) ، قطبش پذیری، و یک بار خالص ثابت ( عموماً از محاسبات کوانتومی و / یا آزمایش تجربی ) گرفته می شود.
• برهم کنش های پیوندی به عنوان فنرهای با فاصله تعادل برابر با طول پیوند تجربی یا محاسبه شده در نظر گرفته می شوند
تنوع پیش زمینه های محاسباتی ممکن در این زمینه زیاد است. به عنوان مثال، بسیاری از شبیه سازی ها در طول تاریخ از یک نمایش اتم واحد استفاده کرده اند که در آن هر ترمینال گروه متیل یا واحد متیلن میانی به عنوان یک ذره در نظر گرفته می شود، و سیستم های پروتئینی بزرگ معمولاً با استفاده از یک مدل تسیح وار یا زنجیر وار شبیه سازی می شوند که دو یا چهار ذره در هر اسید آمینه به یک زنجیر اختصاص می یابد.
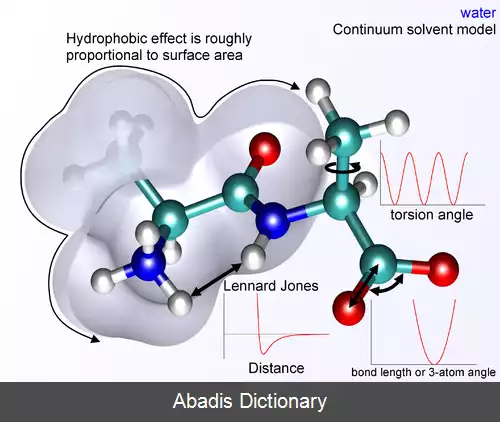
فرض تابعی زیر که به عنوان یک تابع پتانسیل بینا اتمی یا میدان نیرو در شیمی نامیده می شود، انرژی پتانسیل سیستم مولکولی ( E ) را در یک ساختار معین محاسبه می کند که حاصل جمع انرژی عبارات منفرد انرژی است.
E = E covalent + E noncovalent
که در آن اجزای سهم کووالانسی و غیر کووالانسی با تجمیع عبارات منفرد انرژی به شکل زیر آورده شده اند:
E covalent = E bond + E angle + E dihedral
E noncovalent = E electrostatic + E van der Waals
شکل دقیق تابعی از تابع پتانسیل یا میدان نیرو بستگی به برنامه شبیه سازی خاص مورد استفاده دارد. به طور کلی بخش های پیوند و زاویه به عنوان پتانسیل های هارمونیکی حول فاصله تعادلی پیوندی که از محاسبات تجربی ویا تئوری ناشی از ساختار الکترونی با استفاده از نرم افزارهای محاسباتی مانند روش های ابتدا به ساکن مانند Gaussian به دست می آیند مدلسازی می شوند. برای بازتولید دقیق طیفهای ارتعاشی، می توان از پتانسیل مورس با صرف محاسبات رایانه ای استفاده کرد. عبارات زوایای دوسطحی یا پیچشی به طور معمول دارای کمینه های مختلف هستند و بنابراین نمی توانند به عنوان نوسانگر هارمونیک مدل شوند، اگرچه شکل تابعی خاص آنها با پیاده سازی متفاوت است. این کلاس از عبارات ممکن است شامل عبارات نامتناسب دوسطحی ( دوهامنی ) باشد، که بعنوان فاکتورهای تصحیح برای انحراف از سطح عمل می کنند ( به عنوان مثال می توان از آنها برای نگه داشتن مسطح حلقه های بنزن، یا اصلاح هندسه و کایرالی بودن اتم های چهار ضلعی در نمایش اتم واحد استفاده کرد )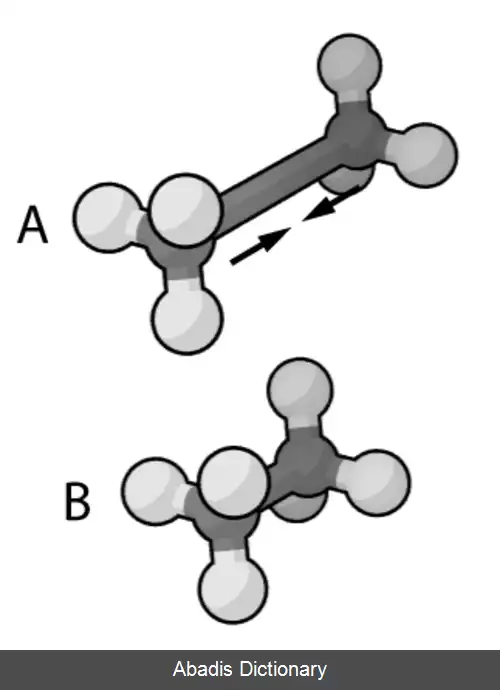
این نوشته برگرفته از سایت ویکی پدیا می باشد، اگر نادرست یا توهین آمیز است، لطفا گزارش دهید: گزارش تخلفروشهای مکانیک مولکولی اتمی دارای خصوصیات زیر است:
• هر اتم به عنوان یک ذره شبیه سازی می شود
• به هر ذره یک شعاع اختصاص داده می شود ( به طور معمول شعاع ون در والس ) ، قطبش پذیری، و یک بار خالص ثابت ( عموماً از محاسبات کوانتومی و / یا آزمایش تجربی ) گرفته می شود.
• برهم کنش های پیوندی به عنوان فنرهای با فاصله تعادل برابر با طول پیوند تجربی یا محاسبه شده در نظر گرفته می شوند
تنوع پیش زمینه های محاسباتی ممکن در این زمینه زیاد است. به عنوان مثال، بسیاری از شبیه سازی ها در طول تاریخ از یک نمایش اتم واحد استفاده کرده اند که در آن هر ترمینال گروه متیل یا واحد متیلن میانی به عنوان یک ذره در نظر گرفته می شود، و سیستم های پروتئینی بزرگ معمولاً با استفاده از یک مدل تسیح وار یا زنجیر وار شبیه سازی می شوند که دو یا چهار ذره در هر اسید آمینه به یک زنجیر اختصاص می یابد.
فرض تابعی زیر که به عنوان یک تابع پتانسیل بینا اتمی یا میدان نیرو در شیمی نامیده می شود، انرژی پتانسیل سیستم مولکولی ( E ) را در یک ساختار معین محاسبه می کند که حاصل جمع انرژی عبارات منفرد انرژی است.
E = E covalent + E noncovalent
که در آن اجزای سهم کووالانسی و غیر کووالانسی با تجمیع عبارات منفرد انرژی به شکل زیر آورده شده اند:
E covalent = E bond + E angle + E dihedral
E noncovalent = E electrostatic + E van der Waals
شکل دقیق تابعی از تابع پتانسیل یا میدان نیرو بستگی به برنامه شبیه سازی خاص مورد استفاده دارد. به طور کلی بخش های پیوند و زاویه به عنوان پتانسیل های هارمونیکی حول فاصله تعادلی پیوندی که از محاسبات تجربی ویا تئوری ناشی از ساختار الکترونی با استفاده از نرم افزارهای محاسباتی مانند روش های ابتدا به ساکن مانند Gaussian به دست می آیند مدلسازی می شوند. برای بازتولید دقیق طیفهای ارتعاشی، می توان از پتانسیل مورس با صرف محاسبات رایانه ای استفاده کرد. عبارات زوایای دوسطحی یا پیچشی به طور معمول دارای کمینه های مختلف هستند و بنابراین نمی توانند به عنوان نوسانگر هارمونیک مدل شوند، اگرچه شکل تابعی خاص آنها با پیاده سازی متفاوت است. این کلاس از عبارات ممکن است شامل عبارات نامتناسب دوسطحی ( دوهامنی ) باشد، که بعنوان فاکتورهای تصحیح برای انحراف از سطح عمل می کنند ( به عنوان مثال می توان از آنها برای نگه داشتن مسطح حلقه های بنزن، یا اصلاح هندسه و کایرالی بودن اتم های چهار ضلعی در نمایش اتم واحد استفاده کرد )


wiki: مکانیک مولکولی
پیشنهاد کاربران
پیشنهادی ثبت نشده است. شما اولین نفر باشید