
فیلترهای بلوم در بیوانفورماتیک ( به انگلیسی: Bloom filters in bioinformatics ) نوعی داده ساختار احتمالاتی هستند که برای ذخیره سازی اعضای یک مجموعه، و جستجو کردن اعضا درون آن مجموعه هستند. هدف استفاده از این طراحی این است که بتوانیم تعدادی از پرس وجوها را با سرعت و به صورت قطعی پاسخ دهیم، و در مواقعی حافظهٔ مورد نیاز را نیز کاهش دهیم.
به عنوان مثال طراحی فیلتر بلوم به این شکل است که اگر پاسخ پرس وجو از فیلتر بلوم منفی باشد، قطعاً عضو مورد نظر در مجموعهٔ متناظر وجود ندارد ( و این را صراحتاً می توانیم اعلام کنیم ) . اما اگر پاسخ پرس وجو مثبت باشد، نمی توان به طور قطع گفت که این عضو در مجموعهٔ مورد نظر وجود دارد یا خیر، و باید عملیات محاسباتی دیگری را برای تأیید یا رد حرف خود به کار بگیریم.
از این داده ساختار، برای بهبود حافظه و سرعت برخی از برنامه های کاربردی در زمینه بیوانفورماتیک به وفور استفاده می شود. در این مقاله، برخی از کاربردهای فیلتر بلوم در حل مسائل بیوانفورماتیک را بررسی می کنیم.
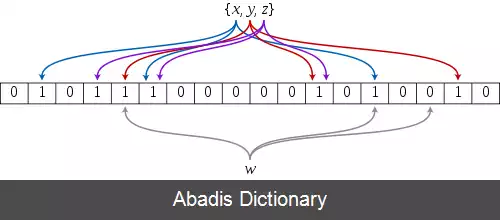
فیلتر بلوم، از تعدادی تابع درهم ساز استفاده می کند و هر تابع، اعضای مجموعه را به یک اندیس از یک آرایه بیتی متناظر می کند. برای اضافه کردن یک عضو به فیلتر بلوم، به ازای هر تابع درهم ساز، آن عضو را Hash می کنیم و اندیس خروجی تابع را در آرایه بیتی، ۱ می کنیم. برای پرس وجو از این فیلتر نیز ورودی را به ازای تمام توابع درهم ساز، Hash کرده و اندیس های متناظر خروجی را با هم AND می کنیم.
مشخص است که اگر عضوی در داده ساختار اضافه شده باشد، تمام بیت های متناظر با اندیس های خروجی توابع، ۱ هستند و در نتیجه خروجی پرسمان نیز مثبت است ( و بالطبع اگر حاصل پرسمان منفی باشد، قطعاً عضو مورد نظر در داده ساختار وجود ندارد ) ؛ اما اگر عضوی در داده ساختار اضافه نشده باشد نیز ممکن است پرسمان خروجی مثبت بدهد ( مثبت کاذب ) . در روش های طراحی فیلتر بلوم، تلاش می شود تا این معیار مثبت کاذب کاهش داده شود.
در تحقیقات بیوانفورماتیک روی ژنوم ها و کارکرد هر قسمت از آن ها، نمی توان یک ژنوم را به طور کامل و یک تکه مورد بررسی قرار داد. به این منظور، ژنوم را به چندین قسمت کوچک تر تقسیم می کنند، و سپس برای یک پارچه سازی، آن ها را کنار هم قرار می دهند.
یکی از پرکاربردترین روش ها در این موضوع، استفاده از گراف دی براین است. در این گراف، به ازای هر k تایی ( k - mer ) از رشته های ژنوم، یک رأس در نظر گرفته می شود، و بین دو رأس A و B در صورتی که یک k − 1 کاراکتر آخر A با k − 1 کاراکتر ابتدای B برابر باشد، یال جهت دار گذاشته می شود. برای کاهش حافظهٔ مصرفی، فیلتر بلوم را به کار می گیریم، و رئوس گراف را در یک فیلتر بلوم نگه می داریم. برای پیمایش گراف نیز از یک رأس، تمام ۴ حالتی که برای کاراکتر آخر رأس بعدی وجود دارد را در فیلتر بلوم پرسمان می کنیم.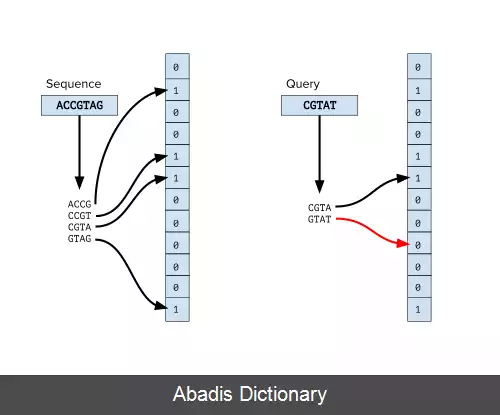
این نوشته برگرفته از سایت ویکی پدیا می باشد، اگر نادرست یا توهین آمیز است، لطفا گزارش دهید: گزارش تخلفبه عنوان مثال طراحی فیلتر بلوم به این شکل است که اگر پاسخ پرس وجو از فیلتر بلوم منفی باشد، قطعاً عضو مورد نظر در مجموعهٔ متناظر وجود ندارد ( و این را صراحتاً می توانیم اعلام کنیم ) . اما اگر پاسخ پرس وجو مثبت باشد، نمی توان به طور قطع گفت که این عضو در مجموعهٔ مورد نظر وجود دارد یا خیر، و باید عملیات محاسباتی دیگری را برای تأیید یا رد حرف خود به کار بگیریم.
از این داده ساختار، برای بهبود حافظه و سرعت برخی از برنامه های کاربردی در زمینه بیوانفورماتیک به وفور استفاده می شود. در این مقاله، برخی از کاربردهای فیلتر بلوم در حل مسائل بیوانفورماتیک را بررسی می کنیم.
فیلتر بلوم، از تعدادی تابع درهم ساز استفاده می کند و هر تابع، اعضای مجموعه را به یک اندیس از یک آرایه بیتی متناظر می کند. برای اضافه کردن یک عضو به فیلتر بلوم، به ازای هر تابع درهم ساز، آن عضو را Hash می کنیم و اندیس خروجی تابع را در آرایه بیتی، ۱ می کنیم. برای پرس وجو از این فیلتر نیز ورودی را به ازای تمام توابع درهم ساز، Hash کرده و اندیس های متناظر خروجی را با هم AND می کنیم.
مشخص است که اگر عضوی در داده ساختار اضافه شده باشد، تمام بیت های متناظر با اندیس های خروجی توابع، ۱ هستند و در نتیجه خروجی پرسمان نیز مثبت است ( و بالطبع اگر حاصل پرسمان منفی باشد، قطعاً عضو مورد نظر در داده ساختار وجود ندارد ) ؛ اما اگر عضوی در داده ساختار اضافه نشده باشد نیز ممکن است پرسمان خروجی مثبت بدهد ( مثبت کاذب ) . در روش های طراحی فیلتر بلوم، تلاش می شود تا این معیار مثبت کاذب کاهش داده شود.
در تحقیقات بیوانفورماتیک روی ژنوم ها و کارکرد هر قسمت از آن ها، نمی توان یک ژنوم را به طور کامل و یک تکه مورد بررسی قرار داد. به این منظور، ژنوم را به چندین قسمت کوچک تر تقسیم می کنند، و سپس برای یک پارچه سازی، آن ها را کنار هم قرار می دهند.
یکی از پرکاربردترین روش ها در این موضوع، استفاده از گراف دی براین است. در این گراف، به ازای هر k تایی ( k - mer ) از رشته های ژنوم، یک رأس در نظر گرفته می شود، و بین دو رأس A و B در صورتی که یک k − 1 کاراکتر آخر A با k − 1 کاراکتر ابتدای B برابر باشد، یال جهت دار گذاشته می شود. برای کاهش حافظهٔ مصرفی، فیلتر بلوم را به کار می گیریم، و رئوس گراف را در یک فیلتر بلوم نگه می داریم. برای پیمایش گراف نیز از یک رأس، تمام ۴ حالتی که برای کاراکتر آخر رأس بعدی وجود دارد را در فیلتر بلوم پرسمان می کنیم.

